Abstract
Dystonia is a network disorder presumed to result from abnormalities in multiple brain regions and in multiple cell populations. The specific pathomechanisms affecting the motor circuits in dystonia are, however, still largely unclear. Animal models for dystonia have long been used to advance our understanding on how specific brain regions and cell populations are involved in dystonia symptomatogenesis. Lesioning, pharmacological modulation and electrical stimulation paradigms were able to highlight that both the basal ganglia and the cerebellum are pathologically altered in these animal models for dystonia. Techniques such as optogenetics and chemogenetics now offer the opportunity for targeted modulation of brain regions and most importantly cell populations and circuits. This could not only allow for a better understanding of the dystonic brain, but potentially improve and expand treatment options. In hopes that the insights from these neuromodulation techniques will eventually translate into therapies, we aim to summarize and critically discuss the findings from different in vivo approaches used to dissect the network dysfunctions underlying dystonia.
Introduction
Dystonia encompasses a heterogeneous group of hyperkinetic movement disorders assumed to be caused by a dysfunctional motor network. The entire cortico-basal ganglia-thalamo-cortical network, the brainstem with regions such as the pedunculopontine nucleus (PPN) as well as cerebellar regions have been implicated in the development of dystonia. The degree to which these brain structures are involved is, however, still unresolved [1, 2].
Further studies are needed to clarify the specific cell populations within these structures causing dystonia. This need is especially pressing since deep brain stimulation (DBS) of the globus pallidus internus (GPi) and the subthalamic nucleus (STN) represent the best therapeutic options for most forms of dystonia. However, the response rate is variable and can be compromised by side effects as well as a secondary failure of DBS [3]. Especially some monogenic forms of dystonia do not respond to GPi or STN DBS. Elucidating the motor circuit changes involved in dystonia development and understanding the mechanisms of action of DBS in dystonia could allow for better neuromodulation protocols and alternative targets.
Over the past decades, different approaches have been utilized in order to study the network pathologies in rodent models for dystonia. Within this review, we will summarize the findings from brain structure lesions in animal models. We will further present the advances in the field of in vivo, region- and cell-specific neuromodulation. Lesioning of brain structures is a common approach in animal models to understand the role of specific brain regions in dystonia symptomatogenesis [4, 5]. DBS in animal models is another method allowing for the modulation of brain nuclei and circuits, albeit with lack of specificity and an underlying mechanistic unclarity [6–9]. Allowing for a more cell-specific intervention of the motor circuit are now optogenetic and chemogenetic tools [10]. For optogenetics, a light-sensitive ion channel is expressed in a specific cell population via targeted injection of a viral vector [10–12]. Light pulses are then applied via an implanted optical fiber, allowing for neuronal depolarization or hyperpolarization of cells. Chemogenetic tools modulate distinct cell populations via engineered receptors or channels, which are selective for specific ligands [13]. This allows for reversible modulation of the cells expressing these receptors or channels. Viral vectors are used in order to express the receptors or channels in the confined neural population. Cell population-specific neuromodulation techniques have not yet been used widely in dystonia basic research. In contrast, for Parkinson’s disease, an optogenetic approach was recently applied to specifically excite parvalbumin-expressing neurons and inhibit lim-homeobox-6–expressing neurons of the globus pallidus externus (GPe) simultaneously in a 6-hydroxydopamine (6-OHDA) mouse model [14]. The authors were able to show a long-lasting effect of stimulation even after DBS was turned off, which could not be seen with conventional DBS. This publication highlights how population-specific neuromodulation can advance our understanding of the circuit defects underlying movement disorders and advance DBS to more targeted stimulation potentially allowing for a better response and less side effects.
Within this review, we will discuss neuromodulation approaches in animal models for dystonia and in some cases for levodopa-induced dyskinesias. It can be assumed that dystonia and levodopa-induced dyskinesias share some common pathomechanisms [15, 16]. This review only touches upon methods of pharmacological modulation, however, does not discuss them at length. Many studies have been performed studying the response of brain regions or cell populations to different drugs [17–19]. We would like to refer to other comprehensive reviews discussing the contributions of these studies [20, 21].
Lesioning of brain regions in dystonia rodent models
Multiple studies have been able to show that dystonia can be elicited in wildtype, non-predisposed animals via lesioning of different brain regions. In wildtype rodents, the injection of 3-nitropropionic acid causes striatal damage and the emergence of a dystonia-like phenotype [22, 23]. Mechanistically, 3-nitropropionic acid primarily leads to a loss of GABAergic striatal projection neurons [24]. In previously healthy baboons, the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) induces transient dystonia-like manifestations [25, 26]. MPTP induces apoptosis in dopaminergic neurons within the substantia nigra. Development of transient dystonia in this model has been reported to be associated with a striatal decrease of dopamine and a transient reduction in D2 dopamine receptors, however, the pathomechanisms have not been clearly elucidated. It has been speculated that abnormal cortico-striatal plasticity might underlie the phenomenon [27]. Both the targeted, pharmacological inactivation as well as activation of the GPi with a GABA agonist or antagonist, respectively, caused dystonic movements of the upper limbs in wildtype monkeys [28, 29]. Contrasting findings such as these have led to the hypothesis that the firing pattern of the GPi might be more relevant in dystonia pathophysiology than the firing rate [16, 30]. Studies in wildtype animals have also identified the cerebellum as a potentially important player in dystonia development. Stimulation of the cerebellum of wildtype mice using injections of the excitatory glutamate agonist kainate led to a dystonia-like phenotype [5, 31, 32]. A hypothesized explanation for this phenomenon is an aberrant firing of Purkinje cells, as dystonia was notably absent in mice without these cells when exposed to kainate [31]. Beyond basal ganglia and cerebellum, pharmacological lesioning of the PPN in wildtype mice triggered dystonia-like behavior in a tail suspension test [33]. In case of bilateral lesions, decreased c-FOS activity, a marker for neuronal activity, was found in the dorsolateral striatum, in the GPi, the STN and the substantia nigra. The PPN is an important interface between the basal ganglia and the cerebellum. Altogether, these studies in non-mutated animal models have highlighted the role of the basal ganglia, the cerebellum and the PPN in dystonia development.
Further underlining these findings are studies in tottering mice, which have a mutation in the Cacna1a gene impairing calcium channel activity and resulting in paroxysmal episodes of stress-induced dystonia-like movements. An aggravation of the dystonia-like phenotype was achieved in tottering mice through lesioning of the striatum by administration of substances such as 6-OHDA or quinolinic acid [5]. It is hypothesized that the manifestation of dystonia in this model predominantly originates from a secondary increased expression of calcium channels within the cerebellum. Indeed, removal of the cerebellum in these mice led to a complete remittance of dystonic attacks [5]. Cerebellectomies were also effective in eliminating dystonic attacks in genetically dystonic rats with a suspected deficiency in the protein caytaxin, which was proposed to be imperative for cerebellar cortex development and function [34, 35]. The cerebellum was also found to underlie development of dystonia in a pharmacological mouse model for DYT/PARK-ATP1A3 dystonia [36]. Blocking the α3-subunit of the sodium-potassium channel with ouabain in the cerebellum led to the development of generalized dystonia-like movements in wildtype mice. The sodium pump dysfunction was shown to be associated with erratic cerebellar activity. In this model, electrical lesions bilaterally ablating the centrolateral thalamus were employed, thus targeting the di-synaptic connection between the cerebellum and the basal ganglia. These lesions yielded a significant reduction in dystonia-like movements. Neurotoxic silencing of the centrolateral thalamus achieved a similar result in the same DYT/PARK-ATP1A3 mouse model, corroborating the findings [37]. Interestingly, bilateral silencing of the motor cortex using the sodium channel blocker tetrodotoxin reduced dystonia only slightly, leading the authors to conclude that the role of the cortex is less essential in DYT/PARK-ATP1A3 pathophysiology than that of the cerebellum [36].
Overall, lesioning of various brain regions has allowed for important strides in understanding whether specific brain structures influence the development of dystonia. Focusing primarily on the basal ganglia and the cerebellum, these lesions have provided insights on the overall importance of these structures in the dystonic network. However, it is important to note that this approach, although valuable, is somewhat imprecise in terms of anatomy and lacks specificity for distinct cell populations. Moreover, it often results in unwanted additional symptoms, such as hypokinesia in case of nigrostriatal damage induced by MPTP or 6-OHDA.
Neuromodulation with DBS in dystonia rodent models
Exploring various animal models of dystonia, researchers have employed DBS targeting the entopeduncular nucleus (EP), which serves as the rodent equivalent of the human GPi. The classical DBS approaches in rodent dystonia models have undergone extensive scrutiny in previous reviews [7, 8]. One study utilized a genetically susceptible DYT-TOR1A rat model and induced dystonia-like movements in the hindlimbs through a peripheral nerve injury [38]. The underlying hypothesis here is the second-hit hypothesis, suggesting the necessity of extragenetic factors to trigger the development of dystonic symptoms in a mutation carrier for dystonia [39, 40]. The authors demonstrated that 3 weeks of high-frequency stimulation effectively alleviated the dystonia-like phenotype and reduced the pathologically enhanced theta power observed in the EP of dystonic animals. An abundance of data has been gathered from DBS of the EP in the dtsz mutant hamster [41–46]. Summarizing the key discoveries, a study revealed circuit plasticity effects following 10 h of unilateral EP-DBS. Both dtsz mutant hamsters and control animals exhibited increased c-Fos expression in the ipsilateral striatum, while dtsz hamsters showed reduced c-Fos expression in the thalamus, distinguishing them from the control group [42]. In vitro measurement of field excitatory postsynaptic potentials from the striatum immediately after 3 h-long high-frequency stimulation in the dtsz mutant hamster revealed effects on cortico-striatal synaptic communication and an increase of the inhibitory tone in the striatal tissue of dystonic animals when compared to non-dystonic hamsters [44].
A study reporting on DBS of the deep cerebellar nuclei and the centrolateral thalamus in dystonia focused on a mouse model targeting the olivocerebellar pathway [47]. A genetic approach was used to specifically silence glutamatergic signaling at the olivocerebellar synapses. The authors conditionally deleted the vesicular glutamate transporter 2 from Ptf1a-expressing excitatory neurons in the inferior olive, thus blocking the fast neuronal communication from climbing fibers to Purkinje cells. Mice with this targeted genetic silencing exhibited a strong dystonia-like phenotype. The loss of climbing fiber neurotransmission was found to cause highly irregular firing of downstream neurons in the cerebellar nuclei. Interestingly, dystonia was alleviated by either silencing the cerebellar nuclei output with lidocaine or by DBS of the interposed nuclei of the cerebellum as well as by DBS of the centrolateral thalamus.
In summary, DBS studies in dystonia animal models have been used to validate the model used, to understand the acute or chronic effects of DBS on different networks and to explore new DBS targets. However, the lack of understanding of the mechanisms of actions of DBS as well as the simultaneous stimulation of more than one cell population can make it difficult to draw conclusions about the underlying neuronal dysfunction. Furthermore, DBS is primarily applied in already symptomatic rodent models, where the network effect of stimulation as well as the effect on the phenotype can be evaluated. However, the most widely used animal model for DBS studies in dystonia is the dystonic hamster dtsz, which is not without its limitations, featuring only transient dystonia and an unidentified genetic mutation [48].
Cell-specific neuromodulation in dystonia rodent models
Only a handful of studies have looked at the cell populations of the striatum in dystonia using optogenetics (Figure 1). Using DYT-TOR1A knock-in mice expressing channelrhodopsin-2 in cholinergic interneurons, Richter et al. applied bilateral optogenetic stimulation to the dorsolateral striatum with the aim of provoking a dystonia-like phenotype [49]. The striatal cholinergic system has long been suspected to play a major role in dystonia. Aside from anticholinergic drugs effectively alleviating symptoms in some dystonia patients, DYT-TOR1A mouse models have shown an abnormal excitation of cholinergic interneurons and increased striatal acetylcholine [50]. By optogenetically stimulating the striatal cholinergic interneurons, the authors aimed to increase the activity of these neurons as well as the acetylcholine levels, thus studying whether this could directly cause dystonia in genetically-predisposed mice. Burst firing with increased release of acetylcholine was optogenetically induced. DYT-TOR1A knock-in mice responded with transient hyperactivity, while wildtype animals did not. On the other hand, in a transgenic mouse model expressing cre-recombinase under the control of the choline acetyltransferase promoter (ChAT-Cre), channelrhodopsin-2 was selectively expressed in cholinergic neurons in order to study the contributions of the cholinergic system to levodopa-induced dyskinesias [51]. Intriguingly, following a unilateral 6-OHDA lesion, the researchers demonstrated that optical pulses targeted at the dorsolateral striatum increased the frequency of levodopa-induced dyskinesias. These observations underline the contribution of cholinergic interneurons to the development of abnormal movement output, but reveal that a disturbance of this population alone is insufficient for dystonia development. Another group of striatal interneurons presumed to play an important role in hyperkinetic movement disorders are parvalbumin-positive, fast-spiking interneurons, which exert a strong feedforward inhibition on medium spiny neurons. In the dtsz hamster model of paroxysmal dystonia, a delayed maturation of parvalbumin-positive interneurons coincides with an age-dependent development of a dystonia-like phenotype and abnormal basal ganglia output [52]. However, although selectively inhibiting striatal parvalbumin-positive interneurons led to a hyperactivation of cholinergic interneurons in DYT-TOR1A knock-in mice compared to wildtype controls, this manipulation failed to induce any behavioral changes, particularly dystonia [53]. Interestingly, optogenetic activation of the striatal D1 medium spiny neurons triggered dyskinesias in 6-OHDA mice [54]. Conversely, the chemogenetic inhibition of D2 medium spiny neurons triggered dyskinesia attacks in a mouse model for paroxysmal non-kinesigenic dyskinesia [55]. So far, the direct and indirect pathways have not been optogenetically targeted in rodent models for dystonia.
FIGURE 1
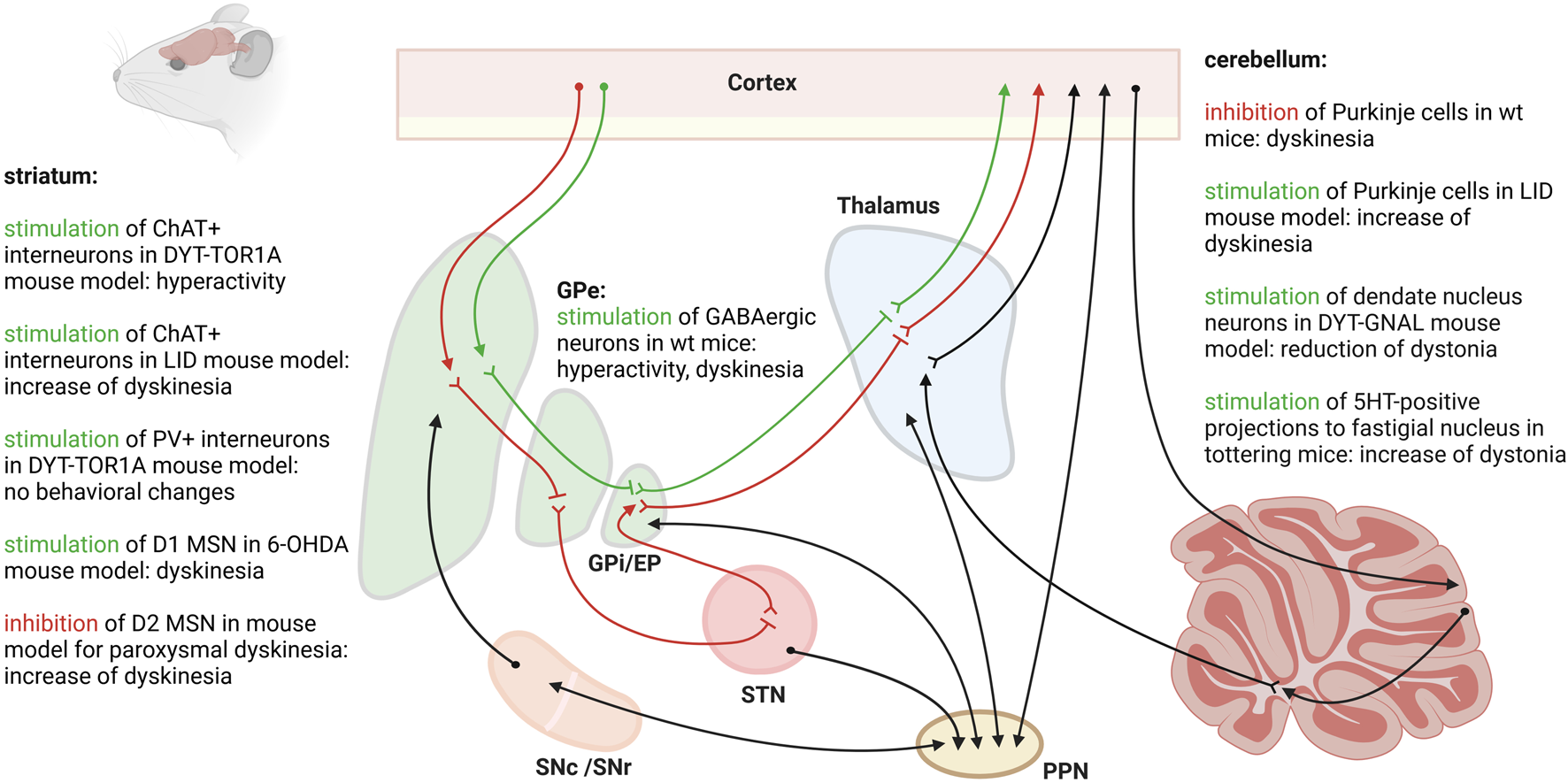
Different cell populations and regions of the central motor circuit have been targeted with optogenetics in rodent models for dystonia or levodopa-induced dyskinesias (LID). The figure was created with biorender.com.
Optogenetic stimulation of the right GPe with targeted expression of ChR2 in the GABAergic neurons led to hyperactivity and dyskinesias with torsion of the neck and of the left forelimb in mice [56]. The authors found that stimulation of the GABAergic neurons of the GPe reduced c-Fos expression in the EP and increased c-Fos expression in the motor cortex and the striatum.
Optogenetic stimulation of the cerebellar cortex in a mouse model expressing ChR2 in molecular layer interneurons was used to locally suppress the activity of Purkinje cells. This inhibition of Purkinje cell activity was shown to induce involuntary movements in mice [57]. Using single-unit extracellular activity recordings, the authors determined that suppression of Purkinje cell activity leads to increased firing, i.e., a disinhibition, of the downstream neurons of the deep cerebellar nuclei of the cerebellum. Conversely, chronic optogenetic stimulation at theta frequency of Purkinje cells expressing ChR2-YFP led to a significant improvement of dyskinesias in a mouse model for levodopa-induced dyskinesias [58]. The effect even outlasted the end of cerebellar stimulation by 2 weeks. The authors found that chronic Purkinje cell stimulation specifically influenced the aberrant firing patterns recorded in the interposed nuclei of the cerebellum, in the motor cortex and in the parafascicular thalamus in mice with levodopa-induced dyskinesias. Specific chemogenetic inactivation of projections from the deep cerebellar nuclei to the parafascicular thalamus prevented the beneficial effect from chronic Purkinje cell stimulation highlighting the importance of the cerebello-thalamic pathway in hyperkinetic movement disorders. Indeed, optogenetic activation of the parafascicular thalamus induced abnormal involuntary movements in wildtype rats [59].
The three nuclei making up the deep cerebellar nuclei are the fastigial nucleus, the interposed nucleus and the dentate nucleus. The neurons of the dentate nucleus were targeted with optogenetic stimulation in an asymptomatic and a symptomatic state in a DYT-GNAL knock-out mouse model [60]. In their asymptomatic state, DYT-GNAL mice revealed increased excitability of the cerebello-thalamic pathway upon low-frequency stimulation of the dentate nucleus compared to wildtype controls. Dystonia-like movements were transiently induced by a nonselective cholinergic agonist, which led to a long-lasting further increase in cerebello-thalamic excitability when stimulation was applied 2 days post drug exposure in DYT-GNAL mice. Theta-burst stimulations of the dentate nucleus were able to reduce cerebello-thalamic excitability and the dystonia-like phenotype in symptomatic DYT-GNAL mice. In tottering mice with stress-induced attacks of dystonia, optogenetic stimulation of the serotonin (5HT)-positive dorse raphei nuclei inputs to the fastigial nucleus led to an increase in the frequency of dystonic attacks [61]. Single-unit recordings revealed an enhanced firing rate of the neurons of the fastigial nucleus upon photostimulation. On the other hand, bilateral photoinhibition of the 5HT-positive inputs to the fastigial nucleus reduced dystonic attacks. Bilateral knockdown of 5HT-2A receptor genes in the fastigial nucleus using short hairpin RNA reduced the frequency of dystonia attacks. The authors conclude that stress increases the excitability of the deep cerebellar nuclei via 5HT, which was directly correlated with the development of transient dystonia.
Taken together, cell population-targeted studies have led to compelling findings such as the involvement of the cerebello-thalamo-striatal pathway in dystonia development and the 5-HT positive projections to the deep cerebellar nuclei as possible targets for DBS [60, 61]. The challenge in targeting single cell populations in dystonia might be the final interpretation of the findings, since dystonia is assumed to be a network disorder with a possible malfunction of multiple structures and multiple cell populations [62].
Discussion
The advent of cell-specific stimulation and inhibition techniques holds great promise in unraveling the intricate involvement of specific neuron populations in dystonia. These techniques hold the potential to pave the way for the exploration of optimized stimulation techniques for DBS in human patients. As exemplified by the study of Spix et al. for Parkinson’s disease, showing advantages of a cell-population specific stimulation in the GPe compared to conventional DBS, the answer to better stimulation paradigms might not necessarily be a different target, but a more specific stimulation technique [14]. On the other hand, in the field of dystonia, there are first encouraging studies exploring new targets such as the optogenetic theta-burst stimulation of dentate nucleus neurons in a symptomatic mouse model for DYT-GNAL [64]. DYT-GNAL is a form of monogenic dystonia known to have a very variable response to GPi DBS in humans [63]. Of course, both optogenetic- and chemogenetic-mediated neuromodulation do have limitations, such as, for example, the need for an invasive application of a viral vector into the target brain structure. Additionally, interpreting the findings in animal studies in dystonia remains an overall challenge due to the difficulties in clearly defining a dystonic phenotype. The scientific community still has not defined a common set of parameters for dystonia manifestation in animal models [4]. However, embracing these advancements is key to moving our understanding and treatment of dystonia forward. Aside from the insights cell specific neuromodulation techniques may give into dystonia pathophysiology in animal models, it has even been proposed that these techniques may eventually be applicable in patients [64, 65].
Statements
Author Contributions
Conceptualization: CI and LR. Writing of the original draft: LR. Review, editing and supervision: CI. All authors contributed to the article and approved the submitted version.
Funding
LR is supported by the Dystonia Medical Research Foundation in Illinois, United States (Postdoctoral Research Fellowship Award) and the Interdisciplinary Center for Clinical Research (IZKF) at the University of Würzburg (ZZ-29). In addition, this project has received funding from the German Research Foundation (DFG) (Project-ID 424778381-TRR 295 A01, A06 to CI) as well as from the European Union’s Horizon 2020 research and innovation program under the EJP RD COFUND-EJP N° 825575 (EurDyscover). Moreover, CI is supported by the Interdisciplinary Center for Clinical Research (IZKF) at the University of Würzburg (A-303, A-421, N-362; S-506) and by the VERUM Foundation.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
Brüggemann N . Contemporary functional neuroanatomy and pathophysiology of dystonia. J Neural Transm (2021) 128(4):499–508. 10.1007/s00702-021-02299-y
2
Lerner RP Niethammer M Eidelberg D . Understanding the anatomy of dystonia: determinants of penetrance and phenotype. Curr Neurol Neurosci Rep (2013) 13(11):401. 10.1007/s11910-013-0401-0
3
Vidailhet M Jutras MF Grabli D Roze E . Deep brain stimulation for dystonia. J Neurol Neurosurg Psychiatry (2013) 84(9):1029–42. 10.1136/jnnp-2011-301714
4
Jinnah HA Hess EJ Ledoux MS Sharma N Baxter MG Delong MR . Rodent models for dystonia research: characteristics, evaluation, and utility. Mov Disord (2005) 20(3):283–92. 10.1002/mds.20364
5
Neychev VK Fan X Mitev VI Hess EJ Jinnah HA . The basal ganglia and cerebellum interact in the expression of dystonic movement. Brain (2008) 131(Pt 9):2499–509. 10.1093/brain/awn168
6
Udupa K Chen R . The mechanisms of action of deep brain stimulation and ideas for the future development. Prog Neurobiol (2015) 133:27–49. 10.1016/j.pneurobio.2015.08.001
7
Knorr S Musacchio T Paulat R Matthies C Endres H Wenger N et al Experimental deep brain stimulation in rodent models of movement disorders. Exp Neurol (2022) 348:113926. 10.1016/j.expneurol.2021.113926
8
Perl S Lüttig A Köhling R Richter A . Deep brain stimulation in animal models of dystonia. Neurobiol Dis (2022) 175:105912. 10.1016/j.nbd.2022.105912
9
Rauschenberger L Güttler C Volkmann J Kühn AA Lofredi R . A translational perspective on pathophysiological changes of oscillatory activity in dystonia and parkinsonism. Exp Neurol (2022) 355:114140. 10.1016/j.expneurol.2022.114140
10
Vlasov K Van Dort CJ Solt K . Optogenetics and chemogenetics. Methods Enzymol (2018) 603:181–96. 10.1016/bs.mie.2018.01.022
11
Deisseroth K . Optogenetics. Nat Methods (2011) 8(1):26–9. 10.1038/nmeth.f.324
12
Deisseroth K . Optogenetics: 10 years of microbial opsins in neuroscience. Nat Neurosci (2015) 18(9):1213–25. 10.1038/nn.4091
13
English JG Roth BL . Chemogenetics-A transformational and translational platform. JAMA Neurol (2015) 72(11):1361–6. 10.1001/jamaneurol.2015.1921
14
Spix TA Nanivadekar S Toong N Kaplow IM Isett BR Goksen Y et al Population-specific neuromodulation prolongs therapeutic benefits of deep brain stimulation. Science (2021) 374(6564):201–6. 10.1126/science.abi7852
15
Calabresi P Standaert DG . Dystonia and levodopa-induced dyskinesias in Parkinson's disease: is there a connection?Neurobiol Dis (2019) 132:104579. 10.1016/j.nbd.2019.104579
16
Scarduzio M Hess EJ Standaert DG Eskow Jaunarajs KL . Striatal synaptic dysfunction in dystonia and levodopa-induced dyskinesia. Neurobiol Dis (2022) 166:105650. 10.1016/j.nbd.2022.105650
17
Maltese M Martella G Madeo G Fagiolo I Tassone A Ponterio G et al Anticholinergic drugs rescue synaptic plasticity in DYT1 dystonia: role of M1 muscarinic receptors. Mov Disord (2014) 29(13):1655–65. 10.1002/mds.26009
18
Sciamanna G Bonsi P Tassone A Cuomo D Tscherter A Viscomi MT et al Impaired striatal D2 receptor function leads to enhanced GABA transmission in a mouse model of DYT1 dystonia. Neurobiol Dis (2009) 34(1):133–45. 10.1016/j.nbd.2009.01.001
19
Sciamanna G Ponterio G Tassone A Maltese M Madeo G Martella G et al Negative allosteric modulation of mGlu5 receptor rescues striatal D2 dopamine receptor dysfunction in rodent models of DYT1 dystonia. Neuropharmacology (2014) 85:440–50. 10.1016/j.neuropharm.2014.06.013
20
Downs AM Roman KM Campbell SA Pisani A Hess EJ Bonsi P . The neurobiological basis for novel experimental therapeutics in dystonia. Neurobiol Dis (2019) 130:104526. 10.1016/j.nbd.2019.104526
21
Jinnah H Richter A Mink JW Caldwell GA Caldwell KA Gonzalez-Alegre P et al Animal models for drug discovery in dystonia. Expert Opin Drug Discov (2008) 3(1):83–97. 10.1517/17460441.3.1.83
22
Palfi S Leventhal L Goetz CG Hantraye T Roitberg BZ Sramek J et al Delayed onset of progressive dystonia following subacute 3-nitropropionic acid treatment in Cebus apella monkeys. Mov Disord (2000) 15(3):524–30. 10.1002/1531-8257(200005)15:3<524::AID-MDS1016>3.0.CO;2-F
23
Fernagut PO Diguet E Stefanova N Biran M Wenning GK Canioni P et al Subacute systemic 3-nitropropionic acid intoxication induces a distinct motor disorder in adult C57Bl/6 mice: behavioural and histopathological characterisation. Neuroscience (2002) 114(4):1005–17. 10.1016/s0306-4522(02)00205-1
24
Beal MF Brouillet E Jenkins BG Ferrante RJ Kowall NW Miller JM et al Neurochemical and histologic characterization of striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. J Neurosci (1993) 13(10):4181–92. 10.1523/JNEUROSCI.13-10-04181.1993
25
Perlmutter JS Tempel LW Black KJ Todd RD . MPTP induces dystonia and parkinsonism. Clues to the pathophysiology of dystonia. Neurology (1997) 49(5):1432–8. 10.1212/wnl.49.5.1432
26
Tabbal SD Mink JW Antenor JAV Carl JL Moerlein SM Perlmutter JS . 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced acute transient dystonia in monkeys associated with low striatal dopamine. Neuroscience (2006) 141(3):1281–7. 10.1016/j.neuroscience.2006.04.072
27
Norris SA Tian L Williams EL Perlmutter JS . Transient dystonia correlates with parkinsonism after 1-methyl-4-phenyl-1,2,3,6- tetrahydropyridine in non-human primates. Dystonia (2023) 2:11019. 10.3389/dyst.2023.11019
28
Burbaud P Bonnet B Guehl D Lagueny A Bioulac B . Movement disorders induced by gamma-aminobutyric agonist and antagonist injections into the internal globus pallidus and substantia nigra pars reticulata of the monkey. Brain Res (1998) 780(1):102–7. 10.1016/s0006-8993(97)01158-x
29
Wenger KK Musch KL Mink JW . Impaired reaching and grasping after focal inactivation of globus pallidus pars interna in the monkey. J Neurophysiol (1999) 82(5):2049–60. 10.1152/jn.1999.82.5.2049
30
Wilson BK Hess EJ . Animal models for dystonia. Mov Disord (2013) 28(7):982–9. 10.1002/mds.25526
31
Pizoli CE Jinnah HA Billingsley ML Hess EJ . Abnormal cerebellar signaling induces dystonia in mice. J Neurosci (2002) 22(17):7825–33. 10.1523/JNEUROSCI.22-17-07825.2002
32
Raike RS Pizoli CE Weisz C van den Maagdenberg AMJM Jinnah HA Hess EJ . Limited regional cerebellar dysfunction induces focal dystonia in mice. Neurobiol Dis (2013) 49:200–10. 10.1016/j.nbd.2012.07.019
33
Su JH Hu YW Song YP Yang Y Li RY Zhou KG et al Dystonia-like behaviors and impaired sensory-motor integration following neurotoxic lesion of the pedunculopontine tegmental nucleus in mice. Front Neurol (2023) 14:1102837. 10.3389/fneur.2023.1102837
34
LeDoux MS Lorden JF Ervin JM . Cerebellectomy eliminates the motor syndrome of the genetically dystonic rat. Exp Neurol (1993) 120(2):302–10. 10.1006/exnr.1993.1064
35
Xiao J Ledoux MS . Caytaxin deficiency causes generalized dystonia in rats. Brain Res Mol Brain Res (2005) 141(2):181–92. 10.1016/j.molbrainres.2005.09.009
36
Calderon DP Fremont R Kraenzlin F Khodakhah K . The neural substrates of rapid-onset Dystonia-Parkinsonism. Nat Neurosci (2011) 14(3):357–65. 10.1038/nn.2753
37
Chen CH Fremont R Arteaga-Bracho EE Khodakhah K . Short latency cerebellar modulation of the basal ganglia. Nat Neurosci (2014) 17(12):1767–75. 10.1038/nn.3868
38
Knorr S Rauschenberger L Pasos UR Friedrich MU Peach RL Grundmann-Hauser K et al The evolution of dystonia-like movements in TOR1A rats after transient nerve injury is accompanied by dopaminergic dysregulation and abnormal oscillatory activity of a central motor network. Neurobiol Dis (2021) 154:105337. 10.1016/j.nbd.2021.105337
39
Rauschenberger L Knorr S Pisani A Hallett M Volkmann J . Second hit hypothesis in dystonia: dysfunctional cross talk between neuroplasticity and environment?Neurobiol Dis (2021) 159:105511. 10.1016/j.nbd.2021.105511
40
Rauschenberger L Krenig EM Stengl A Knorr S Harder TH Steeg F et al Peripheral nerve injury elicits microstructural and neurochemical changes in the striatum and substantia nigra of a DYT-TOR1A mouse model with dystonia-like movements. Neurobiol Dis (2023) 179:106056. 10.1016/j.nbd.2023.106056
41
Harnack D Hamann M Meissner W Morgenstern R Kupsch A Richter A . High-frequency stimulation of the entopeduncular nucleus improves dystonia in dtsz hamsters. Neuroreport (2004) 15(9):1391–3. 10.1097/01.wnr.0000130435.75715.64
42
Reese R Charron G Nadjar A Aubert I Thiolat ML Hamann M et al High frequency stimulation of the entopeduncular nucleus sets the cortico-basal ganglia network to a new functional state in the dystonic hamster. Neurobiol Dis (2009) 35(3):399–405. 10.1016/j.nbd.2009.05.022
43
Leblois A Reese R Labarre D Hamann M Richter A Boraud T et al Deep brain stimulation changes basal ganglia output nuclei firing pattern in the dystonic hamster. Neurobiol Dis (2010) 38(2):288–98. 10.1016/j.nbd.2010.01.020
44
Heerdegen M Zwar M Franz D Hörnschemeyer MF Neubert V Plocksties F et al Mechanisms of pallidal deep brain stimulation: alteration of cortico-striatal synaptic communication in a dystonia animal model. Neurobiol Dis (2021) 154:105341. 10.1016/j.nbd.2021.105341
45
Paap M Perl S Lüttig A Plocksties F Niemann C Timmermann D et al Deep brain stimulation by optimized stimulators in a phenotypic model of dystonia: effects of different frequencies. Neurobiol Dis (2021) 147:105163. 10.1016/j.nbd.2020.105163
46
Gernert M Bennay M Fedrowitz M Rehders JH Richter A . Altered discharge pattern of basal ganglia output neurons in an animal model of idiopathic dystonia. J Neurosci (2002) 22(16):7244–53. 10.1523/JNEUROSCI.22-16-07244.2002
47
White JJ Sillitoe RV . Genetic silencing of olivocerebellar synapses causes dystonia-like behaviour in mice. Nat Commun (2017) 8(1):14912. 10.1038/ncomms14912
48
Löscher W Fisher JE Schmidt D Fredow G Hönack D Iturrian WB . The sz mutant hamster: a genetic model of epilepsy or of paroxysmal dystonia?Mov Disord (1989) 4(3):219–32. 10.1002/mds.870040304
49
Richter F Bauer A Perl S Schulz A Richter A . Optogenetic augmentation of the hypercholinergic endophenotype in DYT1 knock-in mice induced erratic hyperactive movements but not dystonia. EBioMedicine (2019) 41:649–58. 10.1016/j.ebiom.2019.02.042
50
Eskow Jaunarajs KL Bonsi P Chesselet MF Standaert DG Pisani A . Striatal cholinergic dysfunction as a unifying theme in the pathophysiology of dystonia. Prog Neurobiol (2015) 127-128:91–107. 10.1016/j.pneurobio.2015.02.002
51
Bordia T Perez XA Heiss J Zhang D Quik M . Optogenetic activation of striatal cholinergic interneurons regulates L-dopa-induced dyskinesias. Neurobiol Dis (2016) 91:47–58. 10.1016/j.nbd.2016.02.019
52
Hamann M Richter A Meillasson FV Nitsch C Ebert U . Age-related changes in parvalbumin-positive interneurons in the striatum, but not in the sensorimotor cortex in dystonic brains of the dt(sz) mutant hamster. Brain Res (2007) 1150:190–9. 10.1016/j.brainres.2007.02.074
53
Schulz A Richter F Richter A . In vivo optogenetic inhibition of striatal parvalbumin-reactive interneurons induced genotype-specific changes in neuronal activity without dystonic signs in male DYT1 knock-in mice. J Neurosci Res (2023) 101(4):448–63. 10.1002/jnr.25157
54
Ryan MB Bair-Marshall C Nelson AB . Aberrant striatal activity in parkinsonism and levodopa-induced dyskinesia. Cell Rep (2018) 23(12):3438–46. 10.1016/j.celrep.2018.05.059
55
Nelson AB Girasole AE Lee HY Ptáček LJ Kreitzer AC . Striatal indirect pathway dysfunction underlies motor deficits in a mouse model of paroxysmal dyskinesia. J Neurosci (2022) 42(13):2835–48. 10.1523/JNEUROSCI.1614-20.2022
56
Tian J Yan Y Xi W Zhou R Lou H Duan S et al Optogenetic stimulation of GABAergic neurons in the globus pallidus produces hyperkinesia. Front Behav Neurosci (2018) 12:185. 10.3389/fnbeh.2018.00185
57
Heiney SA Kim J Augustine GJ Medina JF . Precise control of movement kinematics by optogenetic inhibition of Purkinje cell activity. J Neurosci (2014) 34(6):2321–30. 10.1523/JNEUROSCI.4547-13.2014
58
Coutant B Frontera JL Perrin E Combes A Tarpin T Menardy F et al Cerebellar stimulation prevents Levodopa-induced dyskinesia in mice and normalizes activity in a motor network. Nat Commun (2022) 13(1):3211. 10.1038/s41467-022-30844-0
59
Chung M Park YS . Hyperkinetic rat model induced by optogenetic parafascicular nucleus stimulation. J Korean Neurosurg Soc (2023) 66(2):121–32. 10.3340/jkns.2022.0106
60
Aïssa HB Sala RW Georgescu Margarint EL Frontera JL Varani AP Menardy F et al Functional abnormalities in the cerebello-thalamic pathways in a mouse model of DYT25 dystonia. Elife (2022) 11:11. 10.7554/elife.79135
61
Kim JE Chae S Kim S Jung YJ Kang MG Heo WD et al Cerebellar 5HT-2A receptor mediates stress-induced onset of dystonia. Sci Adv (2021) 7(10):eabb5735. 10.1126/sciadv.abb5735
62
Jinnah HA Neychev V Hess EJ . The anatomical basis for dystonia: the motor network model. Tremor Other Hyperkinet Mov (N Y) (2017) 7:506. 10.7916/D8V69X3S
63
Tisch S Kumar KR . Pallidal deep brain stimulation for monogenic dystonia: the effect of gene on outcome. Front Neurol (2020) 11:630391. 10.3389/fneur.2020.630391
64
Poth KM Texakalidis P Boulis NM . Chemogenetics: beyond lesions and electrodes. Neurosurgery (2021) 89(2):185–95. 10.1093/neuros/nyab147
65
Chen Y Xiong M Zhang SC . Illuminating Parkinson's therapy with optogenetics. Nat Biotechnol (2015) 33(2):149–50. 10.1038/nbt.3140
Summary
Keywords
dystonia, network disorder, deep brain stimulation, optogenetics, basal ganglia, cerebellum, chemogenetics
Citation
Rauschenberger L and Ip CW (2024) Unraveling dystonia circuitry in rodent models using novel neuromodulation techniques. Dystonia 3:11793. doi: 10.3389/dyst.2024.11793
Received
11 July 2023
Accepted
08 February 2024
Published
19 February 2024
Volume
3 - 2024
Edited by
Jan K. Teller, Scientific Advisors International, Poland
Updates
Copyright
© 2024 Rauschenberger and Ip.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lisa Rauschenberger, rauschenb_l1@ukw.de
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.